GENOMIC
Mapping
11q13.5. View the map and BAC clones (data from UCSC genome browser).
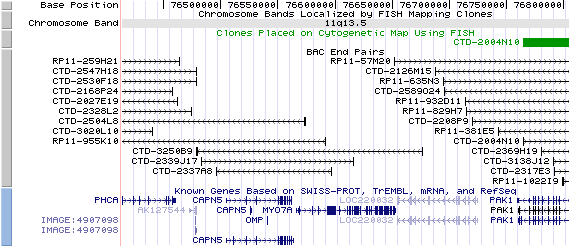
Structure
(assembly 07/03)
MYO7A/NM_000260: 49 exons, 86,974bp, Chr11: 76,565,618-76,652,591.
The figure below shows the structure of the MYO7A gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
MYO7A (NM_000260), 7,465bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: Expressed in the pigment epithelium and the photoreceptor cells of the retina. Also found in kidney, liver, testis, cochlea, lymphocytes. Not expressed in brain.
Detected in optic cup in 5.5 weeks-old embryos. Expressed in retinal pigment epithelium, cochlear and vestibular neuroepithelia, and olfactory epithelium at 8 weeks. At 19 weeks, present in both pigment epithelium and photoreceptor cells. At 24-28 weeks, expression in pigment epithelium and photoreceptor cells increases. Present in pigment epithelium and photoreceptor cells in adult.
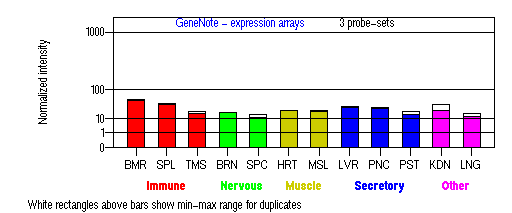
BMR: Bone marrow; SPL: Spleen; TMS: Thymus; BRN: Brain; SPC: Spinal cord; HRT: Heart; MSL: Skeletal muscle; LVR; Liver; PNC: Pancreas; PST: Prostate; KDN: Kidney; LNG: Lung. (data from GeneCards )
PROTEIN
Sequence
Myosin VIIa (NP_000251): 2215aa, ExPaSy NiceProt view of Swiss-Prot:Q13402.
Ortholog
| Species | Mouse | Rat | Zebrafish | Fruitfly | Worm |
| GeneView | Myo7a/sh1 | Myo7a | 252846 | CG7595/ck | hum6 |
| Protein | NP_032689 (2215aa) | NP_703203 (2177aa) | NP_694515 (2179aa) | NP_523571 (2167aa) | NP_508420 (2098aa) |
| Identities | 96% /2136aa | 94% /2094aa | 83% /1855aa | 61% /1365aa | 49% /1107aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) MYSc 59 - 742
b) IQ regions 743 - 765; 766 - 788; 812 - 834; 835 - 857
c) coiled coil 866 - 935
d) low complexity 979 - 996
e) MyTH4 1017 - 1253; 1747 - 1896
f) B41 1254 - 1469; 1898 - 2115
g) SH3 1606 - 1671
(2) Pfam domains: Pfam:MyTH4; Pfam:SH3; Pfam:IQ; Pfam:Myosin_head.
Graphic view of the Pfam domain structure.
(3) CDD domains:
a) COG0164:myosin class I heavy chain
b) KOG4229: Myosin VII, myosin IXB and related myosins [Cell motility]
(4) Graphic view of InterPro domain structure.
(5) Transmembrane domains predicted by SOSUI: None.
Motif/Site
(1) Predicted results by ScanProsite:
a) IQ motif profile :
767 - 796: score=8.078,
813 - 842: score=7.712,
839 - 863: score=8.352
b) FERM domain profile :
1258 - 1605: score=33.260
1902 - 2205: score=40.584
c) Src homology 3 (SH3) domain profile :
1603 - 1672: score=9.536
d) Tyrosine kinase phosphorylation site : [occurs frequently]
88 - 95: Ryr.DhliY,
244 - 252: RqalDernY,
1453 - 1461: KvkeDvvsY,
2045 - 2052: KfeeDks.Y.
e) cAMP- and cGMP-dependent protein kinase phosphorylation site : [occurs frequently]
146 - 149: KRnS,
604 - 607: RKrS,
1103 - 1106: KKsS,
1117 - 1120: KKkS,
1602 - 1605: RKrS.
f) ATP/GTP-binding site motif A (P-loop) : [occurs frequently]
158 - 165: GesgaGKT
g) Amidation site : [occurs frequently]
972 - 975: rGRR,
1078 - 1081: lGKK.
h) Cell attachment sequence : [occurs frequently]
1659 - 1661: RGD
i) Tyrosine sulfation site : [occurs frequently]
470 - 484:
fkleqeeYdlesidw,
1712 - 1726:
ytleefsYdyfrppp.
j) Bipartite nuclear targeting sequence : [occurs frequently]
1103 - 1119:
KKssvrhklvhltlkkk.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase matched entries found, results here.
(2) ModBase predicted comparative 3D structure of Q13402 from UCSC Genome Sorter.
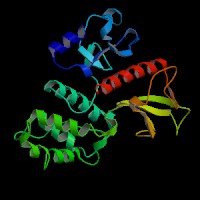
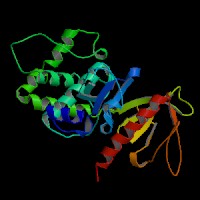
From left to right: Front, Top, and Side views of predicted protein.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=254,406Da, pI=8.78 (NP_000251).
FUNCTION
Ontology
a) Biological processes: visual perception; perception of sound
b) Biological process: actin filament-based movement
c) ATPase activity, coupled
d) A plus-directed microfilament motor activity
e) Component of myosin
f) Component of cytoskeleton
g) Actin binding; ATP binding; calmodulin binding.
Location
Cytoplasmic (probable). In the photoreceptor cells, mainly localized in the inner and base of outer segments as well as in the synaptic ending region.
Interaction
Myosin-VIIa may homodimerize in a two headed molecule through the formation of a coiled-coil rod to consist of two heavy chains, each containing a neck domain with five tandem IQ motifs that bind calmodulins. Myosin VIIa also binds F-actin and shows Mg2+-ATPase activity.
The Rab27a recruits distinct myosins (myosin-VIIa or myosin-Va) via different effectors (MyRIP or melanophilin) to regulate the dynamics of the melanosomes in different cell types (retinal pigment epithelium (RPE) or melanocytes). The melanophilin homolog, MyRIP, mediates the formation of a tripartite complex with RAB27A and myosin VIIa in RPE, to regulate the melanosome distribution (El-Amraoui, et al; Futter, et al). (view diagram of the Myo7a tripartite protein complex in RPE here).
{kind=link}
MyRIP contains a putative actin-binding domain near the C-terminus. Rab27a and MyRIP link neuroendocrine secretory granules to F-actin and control their motion toward release sites through the actin cortex (Desnos, et al). However, the RAB27A/MyRIP protein complex does not appear to require recruitment of a myosin on the secretory granules in PC12 cells for function. Myosin VIIa is apparently not expressed in pancreatic beta cells (Waselle, et al).
Harmonin, the protein encoded by Ush1c, has been shown to bind, by means of its PDZ-domains, with the products of other Usher syndrome type 1 genes, including Myo7a, Cdh23 and Sans. The complexes formed by these protein interactions are thought to be essential for maintaining the integrity of hair cell stereocilia (Johnson, et al; Weil, et al (2003)).
PCDH15 binds to MYO7A and that both proteins are expressed in an overlapping pattern in hair bundles. PCDH15 and MYO7A cooperate to regulate the development and function of the mechanically sensitive hair bundle (Senften, et al).
No entry of the MYO7A drosophila homolog CG7595/ck shown in CuraGen interaction database.
Pathway
In retina, myosin VIIa might play a role in trafficking of ribbon-synaptic vesicle complexes and renewal of the outer photoreceptors disks. Myosin VIIa is involved in the transport of melanosomes along RPE apical processes, similarly to myosin Va in skin melanocytes (Liu, et al (1998)). Myo7a is required for the normal processing of ingested disk membranes in the RPE, primarily in the basal transport of phagosomes into the cell body where they then fuse with lysosomes (Gibbs, et al).
In inner ear, it might maintain the rigidity of stereocilia during the dynamic movements of the bundle. Myosin VIIA participates in anchoring and holding membrane-bound elements to the actin core of the stereocilium. Myosin VIIA is therefore required for the normal gating of transducer channels (Kros, et al).
Involved in hair-cell vesicle trafficking of aminoglycosides, which are known to induce ototoxicity (Richardson, et al). Myosin VIIA is a novel protein kinase A-anchoring protein.
MUTATION
Allele or SNP
Mutational alleles deposited in HGMD.
SNPs deposited in dbSNP.
15 selected allelic examples described in OMIM.
Distribution
80 mutations (1 DFNA11 mutation, 4 DFNB2 mutations, and 75 USH1B mutations) are described (dated on 09/18/2002) in the Deafness Gene Mutation Database at Harvard Medical School Center for Hereditary Deafness.
37 mutations are described in the muation databases of Retina-International scientific Newsletter.
Novel mutations not in the databases:
(1) A novel mutation of the MYO7A gene was reported due to an insertion mutation c.2663_2664insA in a homozygous state in a four generation Indian family, resulting in truncation of MYO7A protein ( Kumar, et al).
(2) Two novel mutations c.397C > G (His133Asp) and 1244-2A > G (Glu459Stop) were found in the MYO7A gene ( Maubaret , et al).
(3) The N458I mutation is described in a family with DFNA11 mutation ( Luijendijk, et al).
(4) A new missense mutation resulting in an Ala230Val change in the motor domain of the myosin VIIA was found in a large Italian family with an autosomal dominant form of non-syndromic sensorineural hearing loss with vestibular involvement ( Di Leva, et al).
(5) Jaijo, et al reported 12 novel mutations of MYO7A in Spamish Usher syndrome type I patients, including five missense mutations, three premature stop codons, three frameshift, and one putative splice-site mutation.
(6) A novel homozygous c.1687G>A mutation in the last nucleotide of exon 14 was reported in a Moroccan USH1B family,and the p.Y1719C represents a frequent polymorphism ( Boulouiz, et al).
(7) Three new myosin VIIA (MYO7A) mutations: p.K923AfsX8, p.Q1896X, and p.E1349K were reported in two Finnish USH1B families( Vastinsalo, et al).
(8) A novel three-nucleotide deletion of MYO7A (p.E1716del) encoding a region of the tail domain was reported by Riazuddin, et al.
Note: Baux, et al have developed a UMD-USHbases database to collect the known mutational alleles and SNPs of the MYO7A gene.
Effect
There are a wide variety of mutations in the MYO7A gene without apparent hotspots. Most of the mutations are missense or nonsense mutations which are predicted to disrupt the function of myosin VIIA protein.
PHENOTYPE
Defects in MYO7A are the cause of Usher syndrome type 1B (USH1B) (Weil, et al (1995)) (OMIM:276903), also known as Usher syndrome non-Acadian variety. Usher syndrome type 1 is an autosomal recessive disease characterized by profound congenital sensorineural deafness, constant vestibular dysfunction and prepubertal onset of progressive retinitis pigmentosa leading to blindness. Usher syndrome is a genetic heterogeneous disorder and the most common cause of combined deafness and blindness in developed countries. Usher syndrome type 1 (USH1) is the most frequent cause of hereditary deaf-blindness in humans. Seven genetic loci (USH1A~G) have been implicated in this disease to date, and five of the corresponding genes have been identified: USH1B, C, D, F, and G. USH1B appears as a primary cytoskeletal protein defect. Patients with USH2 have moderate to severe hearing impairment, normal vestibular function, and later onset of retinal degeneration. USH3 is characterized by progressive hearing loss and variable age of onset of retinal degeneration.
Defects in MYO7A are the cause of autosomal recessive neurosensory deafness 2 (DFNB2) (OMIM:600060), also called neurosensory nonsyndromic recessive deafness 2 (NSRD2). DFNB2 is a form of neurosensory deafness with variable vestibular dysfunction and variable age of onset. (Liu, et al (1997a); Weil, et al (1997)).
Defects in MYO7A are the cause of autosomal dominant nonsyndromic sensorineural deafness 11 (DFNA11) (Liu, et al (1997b); Tamagawa, et al) (OMIM:601317). DFNA11 is a form of nonsyndromic sensorineural deafness with onset after complete speech acquisition and subsequent gradual progression.
REFERENCE
- Baux D, Faug��re V, Larrieu L, Le Gu��dard-M��reuze S, Hamroun D, B��roud C, Malcolm S, Claustres M, Roux AF. UMD-USHbases: a comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum Mutat. 2008; [Epub ahead of print]PMID: 18484607
- Boulouiz R, Li Y, Abidi O, Bolz H, Chafik A, Kubisch C, Roub H, Wollnik B, Barakat A. Analysis of MYO7A in a Moroccan family with Usher syndrome type 1B: novel loss-of-function mutation and non-pathogenicity of p.Y1719C. Mol Vis 2007; 13:1862-5.PMID: 17960123
- Desnos C, Schonn JS, Huet S, Tran VS, El-Amraoui A, Raposo G, Fanget I, Chapuis C, Menasche G, de Saint Basile G, Petit C, Cribier S, Henry JP, Darchen F. Rab27A and its effector MyRIP link secretory granules to F-actin and control their motion towards release sites. J Cell Biol 2003; 163: 559-70. PMID: 14610058
- Di Leva F, D'Adamo P, Cubellis MV, D'Eustacchio A, Errichiello M, Saulino C, Auletta G, Giannini P, Donaudy F, Ciccodicola A, Gasparini P, Franze A, Marciano E. Identification of a Novel Mutation in the Myosin VIIA Motor Domain in a Family with Autosomal Dominant Hearing Loss (DFNA11). Audiol Neurootol 2006; 11: 157-164 PMID: 16449806
- El-Amraoui A, Schonn JS, Kussel-Andermann P, Blanchard S, Desnos C, Henry JP, Wolfrum U, Darchen F, Petit C. MyRIP, a novel Rab effector, enables myosin VIIa recruitment to retinal melanosomes. EMBO Rep 2002; 3: 463-70. PMID: 11964381
- Futter CE, Ramalho JS, Jaissle GB, Seeliger MW, Seabra MC. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Mol Biol Cell 2004; 15: 2264-75. PMID: 14978221
- Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci U S A 2003 ; 100: 6481-6. PMID: 12743369
- Jaijo T, Aller E, Oltra S, Beneyto M, Najera C, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D, Moreno F, Vilela C, Perez-Garrigues H, Navea A, Millan JM. Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum Mutat 2006; 27: 290-1. PMID: 16470552
- Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet 2003; 12: 3075-86. PMID: 14519688
- Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci 2002; 5: 41-7. PMID: 11753415
- Kumar A, Babu M, Kimberling WJ, Venkatesh CP. Genetic analysis of a four generation Indian family with Usher syndrome: a novel insertion mutation in MYO7A. Mol Vis. 2004; 10:910-6. PMID: 15592175
- Liu X, Ondek B, Williams DS. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat Genet 1998; 19: 117-8. (No Abstract)
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet 1997a; 16: 188-90. PMID: 9171832
- Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet 1997b; 17: 268-9. (No Abstract)
- Luijendijk MW, Van Wijk E, Bischoff AM, Krieger E, Huygen PL, Pennings RJ, Brunner HG, Cremers CW, Cremers FP, Kremer H. Identification and molecular modelling of a mutation in the motor head domain of myosin VIIA in a family with autosomal dominant hearing impairment (DFNA11). Hum Genet 2004; 115: 149-56. PMID: 15221449
- Maubaret C, Griffoin JM, Arnaud B, Hamel C. Novel mutations in MYO7A and USH2A in Usher syndrome. Ophthalmic Genet 2005; 26: 25-9.PMID: 15823922
- Riazuddin S, Nazli S, Ahmed ZM, Yang Y, Zulfiqar F, Shaikh RS, Zafar AU, Khan SN, Sabar F, Javid FT, Wilcox ER, Tsilou E, Boger ET, Sellers JR, Belyantseva IA, Riazuddin S, Friedman TB. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat 2008; 29: 502-11. PMID: 18181211
- Richardson GP, Forge A, Kros CJ, Fleming J, Brown SD, Steel KP. Myosin VIIA is required for aminoglycoside accumulation in cochlear hair cells. J Neurosci 1997; 17: 9506-19. PMID: 9391006
- Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Geleoc GS, Gillespie PG, Williams D, Holt JR, Muller U. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci 2006; 26: 2060-71. PMID: 16481439
- Tamagawa Y, Ishikawa K, Ishikawa K, Ishida T, Kitamura K, Makino S, Tsuru T, Ichimura K. Phenotype of DFNA11: a nonsyndromic hearing loss caused by a myosin VIIA mutation. Laryngoscope 2002; 112: 292-7. PMID: 11889386
- Vastinsalo H, Isosomppi J, Aittakorpi A, Sankila EM. Two Finnish USH1B patients with three novel mutations in myosin VIIA. Mol Vis 2006; 12:1093-7. PMID: 17093394
- Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell 2003; 14: 4103-13. PMID: 14517322
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995; 374: 60-1. PMID: 7870171
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 1997; 16:191-3. PMID: 9171833
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003; 12: 463-71. PMID: 12588794
EDIT HISTORY:
Created by Wei Li & Jonathan Bourne: 07/22/2004
Updated by Wei Li: 06/25/2005
Updated by Wei Li: 04/06/2006
Updated by Wei Li: 02/28/2008
Updated by Wei Li, 08/20/2008