GENOMIC
Mapping
22q13.1. View the map and BAC clones (data from UCSC genome browser).
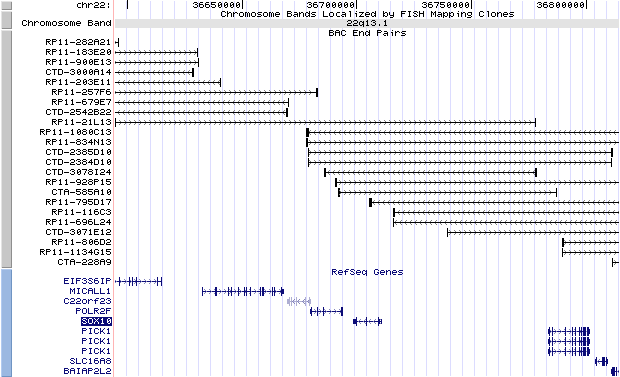
Structure
(assembly 03/2006)
SOX10 (NM_006941): 4 exons, 12,220 bp, chr22:36698266-36710485.
The figure below shows the structure of the SOX10 gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (seq1=mouse Sox10, seq2=human SOX10) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
SOX10( NM_006941), 2,882 bp, view ORF and the alignment to genomic.
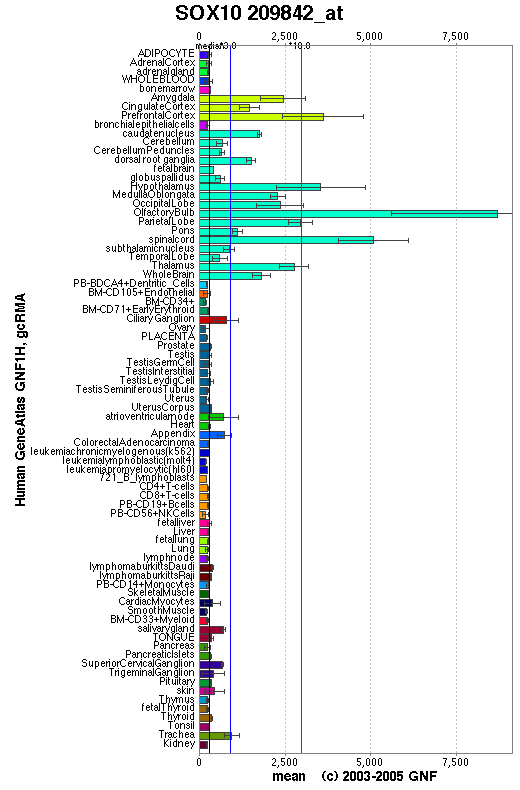
Expression Pattern
Expressed in fetal brain and in adult brain, heart, small intestine and colon.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
View more information in Entrez Gene, or UCSC Gene Sorter, or GeneCards.
PROTEIN
Sequence
SRY (sex determining region Y)-box 10 (NP_008872): 466 aa, UniProtKB/Swiss-Prot entry P56693.
Ortholog
Species Chimpanzee Dog Mouse Rat Chicken GeneView
SOX10
LOC481258
Sox10
Sox10
SOX-10
Protein
XP_525590 (302 aa)
XP_538379 (621 aa)
XP_128139 (569 aa)
NP_062066 (466 aa)
NP_990123 (461 aa)
Identities
281/283 (99%) 455/468 (97%) 459/466 (98%) 454/466 (97%) 388/470 (82%)
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc. View evolutionary tree by TreeView.
Domain
(1) Domains predicted by SMART:
a) low complexity: 23-49
b) HMG: 103-173
c) low complexity: 238-245
d) low complexity: 310-332
(2) Transmembrane domains predicted by SOSUI:
This amino acid sequence is of a SOLUBLE PROTEIN.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a)N-glycosylation site:
Site : 146 to 149 NESD.
b) Protein kinase C phosphorylation site:
Site : 148 to 150 SDK.
Site : 244 to 246 TPK.
Site : 251 to 253 SGK.
Site : 431 to 433 SQR.
c) Casein kinase II phosphorylation site:
Site : 8 to 11 SEVE.
Site : 81 to 84 SGYD.
Site : 262 to 265 SMGE.
Site : 287 to 290 SNME.
Site : 453 to 456 THWE.
d) Tyrosine kinase phosphorylation site:
Site : 166 to 173 KDHPDYKY.
e) N-myristoylation site:
Site : 26 to 31 GSAPSL.
Site : 35 to 40 GGGGGS.
Site : 36 to 41 GGGGSG.
Site : 37 to 42 GGGSGL.
Site : 96 to 101 GASKSK.
Site : 198 to 203 GGTAAI.
Site : 220 to 225 GSPMSD.
Site : 319 to 324 GLGSAL.
Site : 321 to 326 GSALAV.
Site : 416 to 421 GQASGL.
Site : 420 to 425 GLYSAF.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) Tentative number of TMS(s) for the threshold 0.5: 0
c) KDEL ER retention motif in C-terminus: none
d) ER membrane retention signals: none
e) VAC possible vacuolar targeting motif: none
f) Actinin-type actin-binding motif: type 1: none; type 2: none
g) Prenylation motif: none
h) memYQRL transport motif from cell surface to Golgi: none
i) Tyrosines in the tail: none
j) Dileucine motif in the tail: none
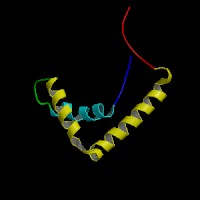
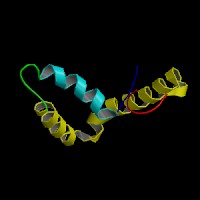
3D Model
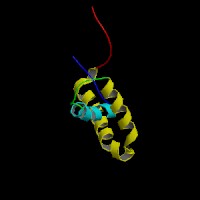
(1)
ModBase predicted 3D structure of P56693 from UCSC Genome Sorter:
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=49911.1Da, pI=6.19.
| Species | Chimpanzee | Dog | Mouse | Rat | Chicken |
| GeneView | SOX10 | LOC481258 | Sox10 | Sox10 | SOX-10 |
| Protein | XP_525590 (302 aa) | XP_538379 (621 aa) | XP_128139 (569 aa) | NP_062066 (466 aa) | NP_990123 (461 aa) |
| Identities | 281/283 (99%) | 455/468 (97%) | 459/466 (98%) | 454/466 (97%) | 388/470 (82%) |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc. View evolutionary tree by TreeView.
Domain
(1) Domains predicted by SMART:
a) low complexity: 23-49
b) HMG: 103-173
c) low complexity: 238-245
d) low complexity: 310-332
(2) Transmembrane domains predicted by SOSUI:
This amino acid sequence is of a SOLUBLE PROTEIN.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a)N-glycosylation site:
Site : 146 to 149 NESD.
b) Protein kinase C phosphorylation site:
Site : 148 to 150 SDK.
Site : 244 to 246 TPK.
Site : 251 to 253 SGK.
Site : 431 to 433 SQR.
c) Casein kinase II phosphorylation site:
Site : 8 to 11 SEVE.
Site : 81 to 84 SGYD.
Site : 262 to 265 SMGE.
Site : 287 to 290 SNME.
Site : 453 to 456 THWE.
d) Tyrosine kinase phosphorylation site:
Site : 166 to 173 KDHPDYKY.
e) N-myristoylation site:
Site : 26 to 31 GSAPSL.
Site : 35 to 40 GGGGGS.
Site : 36 to 41 GGGGSG.
Site : 37 to 42 GGGSGL.
Site : 96 to 101 GASKSK.
Site : 198 to 203 GGTAAI.
Site : 220 to 225 GSPMSD.
Site : 319 to 324 GLGSAL.
Site : 321 to 326 GSALAV.
Site : 416 to 421 GQASGL.
Site : 420 to 425 GLYSAF.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) Tentative number of TMS(s) for the threshold 0.5: 0
c) KDEL ER retention motif in C-terminus: none
d) ER membrane retention signals: none
e) VAC possible vacuolar targeting motif: none
f) Actinin-type actin-binding motif: type 1: none; type 2: none
g) Prenylation motif: none
h) memYQRL transport motif from cell surface to Golgi: none
i) Tyrosines in the tail: none
j) Dileucine motif in the tail: none
3D Model
(1) ModBase predicted 3D structure of P56693 from UCSC Genome Sorter:
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=49911.1Da, pI=6.19.
FUNCTION
Ontology
(1) Biological process: DNA binding, DNA-dependent regulation of transcription, sensory perception of sound.
(2) Melanocyte differentiation.
(3) Gut morphogenesis.
(4) Enteric and peripheral nervous system development.
Location
Nucleus/Cytoplasm
Interaction
Transcription factor SOX-10. Transcription factor that seems to function synergistically with the POU domain protein TST-1/OCT6/SCIP. This gene encodes a member of the SOX (SRY-related HMG-box) family of transcription factors involved in the regulation of embryonic development and in the determination of the cell fate. The encoded protein may act as a transcriptional activator after forming a protein complex with other proteins.
Interactions in HPRD
View co-occured partners in literature searched by PPI Finder.
Pathway
SRY related HMG box gene 10,expressed in neural crest cells during early development and in glial cells of the peripheral and central nervous system during late development and in adult, playing an essential role in the development of neural-crest-derived human cell lineages, homolog to mouse dominant megacolon (Dom).
MUTATION
Allele or SNP
25 mutations deposited in HGMD.
SNPs deposited in dbSNP.
Selected allelic examples described in OMIM.
Distribution
Except the mutations deposited in HGMD, other novel mutations are listed here.
Sznajer, et al (2008) reported a novel missense mutation of SOX10(c.521A>C) in a WS4 patient.
Barnett, et al (2009) reported a novel missense mutation of the HMG domain of SOX10(Q174P; c.521A>C). This case represents the first description of aplasia of the cochlear nerve due to a SOX10 mutation.
Note that the numbering of the mutations in HGMD is based on SNAI2 cDNA (NM_006941), view ORF here.
Effect
The vast majority of disease-associated SOX10 mutations result in premature termination codons (PTCs), causing either WS4 or PCWH depending on the position of the mutations. SOX10 extension mutation (1400del12) is distinct from that of more common PTC mutations. Failure to properly terminate SOX10 translation causes the generation of a deleterious functional domain (WR domain) that occurs because of translation of the normal 3'-UTR (additional 82 aa). The mutant fusion protein causes a severe neurological disease, possibly mediated by a gain-of-function effect (Inoue, et al).
PHENOTYPE
Defects in SOX10 are the cause of Waardenburg syndrome type 2E (WS2E) [MIM:611584]. Waardenburg syndrome (WS; deafness with pigmentary abnormalities) is a congenital disorder caused by defective function of the embryonic neural crest. Depending on additional symptoms, WS is classified into four types: WS1, WS2, WS3 and WS4. WS2 is characterized by deafness and pigmentation defects without additional features. The association of facial dysmorphic features defines WS1 and WS3, whereas the association with Hirschsprung disease (aganglionic megacolon) characterizes WS4, also called "Waardenburg-Hirschsprung disease." Mutations within the genes MITF and SNAI2 have been identified in WS2, whereas mutations of EDN3, EDNRB, and SOX10 have been observed in patients with WS4. SOX10 deletions were found in WS2 cases, making SOX10 a new gene of WS2 (Bondurand, et al).
Defects in SOX10 are a cause of Waardenburg syndrome type 4 (WS4) [MIM:277580]; also known as Waardenburg-Shah syndrome. WS4 is characterized by the association of Waardenburg features (depigmentation and deafness) and the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease).
Defects in SOX10 are a cause of Yemenite deaf-blind hypopigmentation syndrome [MIM:601706]. The disorder consists of cutaneous hypopigmented and hyperpigmented spots and patches, microcornea, coloboma and severe hearing loss. Another case observed in a girl with similar skin symptoms and hearing loss but without microcornea or coloboma is reported as a mild form of this syndrome.
Defects in SOX10 are the cause of peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease (PCWH) [MIM:609136]; also called neurologic variant of Waardenburg-Shah syndrome. PCWH is a rare, complex and more severe neurocristopathy that includes features of 4 distinct syndromes: peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease.
SOX10 gene is related to the development of schizophrenia in the Japanese population (Maeno, et al).
REFERENCE
- Barnett CP, Mendoza-Londono R, Blaser S, Gillis J, Dupuis L, Levin AV, Chiang PW, Spector E, Reardon W. Aplasia of cochlear nerves and olfactory bulbs in association with SOX10 mutation. Am J Med Genet A 2009; 149A:431-6.PMID: 19208381
- Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet 2007; 81:1169-85. PMID: 17999358
- Inoue K, Ohyama T, Sakuragi Y, Yamamoto R, Inoue NA, Yu LH, Goto Y, Wegner M, Lupski JR. Translation of SOX10 3' untranslated region causes a complex severe neurocristopathy by generation of a deleterious functional domain. Hum Mol Genet 2007; 16:3037-46. PMID: 17855451
- Maeno N, Takahashi N, Saito S, Ji X, Ishihara R, Aoyama N, Branko A, Miura H, Ikeda M, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, Iwata N, Inada T, Ozaki N. Association of SOX10 with schizophrenia in the Japanese population. Psychiatr Genet 2007; 17:227-231.PMID: 17621166
- Sznajer Y, Coldea C, Meire F, Delpierre I, Sekhara T, Touraine RL. A de novo SOX10 mutation causing severe type 4 Waardenburg syndrome without Hirschsprung disease. Am J Med Genet A 2008; 146A:1038-41. PMID: 18348267