GENOMIC
Mapping
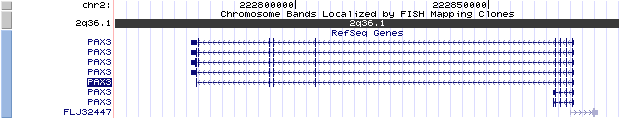
2q36.1. View the map and BAC clones (data from UCSC genome browser).

Structure
(assembly 03/2006)
isoform PAX3 (NM_181457): 10 exons, 99,094 bp, chr2:222772851-222871944.
isoform PAX3a (NM_000438): 4 exons, 5,352 bp, chr2:222866593-222871944.
isoform PAX3b (NM_013942): 5 exons, 5,352 bp, chr2:222866593-222871944.
isoform PAX3d (NM_181458): 9 exons, 99,094 bp, chr2:222772851-222871944.
isoform PAX3e (NM_181459): 10 exons, 99,094 bp, chr2:222772851-222871944.
isoform PAX3g (NM_181461): 8 exons, 97,650 bp, chr2:222774295-222871944.
isoform PAX3h (NM_181460): 9 exons, 99,094 bp, chr2:222772851-222871944.
Note: Seven alternatively spliced isoforms occur: PAX3a, PAX3b, PAX3c (i.e. PAX3), PAX3d, PAX3e,PAX3g, and PAX3h. PAX3a and PAX3b are composed of exons 1 to 4, truncated prematurely in intron 4, and lack the homeodomain and the carboxyl-terminal transactivation domain. PAX3c retains intron 8 and translation continues from exon 8 for five codons into intron 8 before termination. PAX3d lacks intron 8 and translation proceeds from exon 8 to 9. PAX3d is functionally similar to PAX3c. PAX3e contains exons 8, 9, and 10 but lacks introns 8 and 9. PAX3g and PAX3h are truncated isoforms of PAX3d and PAX3e, respectively; both lack part of the transactivation domain encoded by exon 8.
The figure below shows the structure of the PAX3 gene (data from UCSC genome browser).
Regulatory Element
Search the 5'UTR and 1kb upstream regions (seq1=mouse Pax3, seq2=human PAX3) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
a) isoform PAX3 ( NM_181457), 3,193 bp, view ORF and the alignment to genomic.
b) isoform PAX3a ( NM_000438), 1,489 bp, view ORF and the alignment to genomic.
c) isoform PAX3b ( NM_013942), 1,080 bp, view ORF and the alignment to genomic.
d) isoform PAX3d (NM_181458), 3,341 bp, view ORF and the alignment to genomic.
e) isoform PAX3e (NM_181459), 3,170 bp, view ORF and the alignment to genomic.
f) isoform PAX3g (NM_181461), 1,650 bp, view ORF and the alignment to genomic.
g) isoform PAX3h (NM_181460), 2,923 bp, view ORF and the alignment to genomic.
Expression Pattern
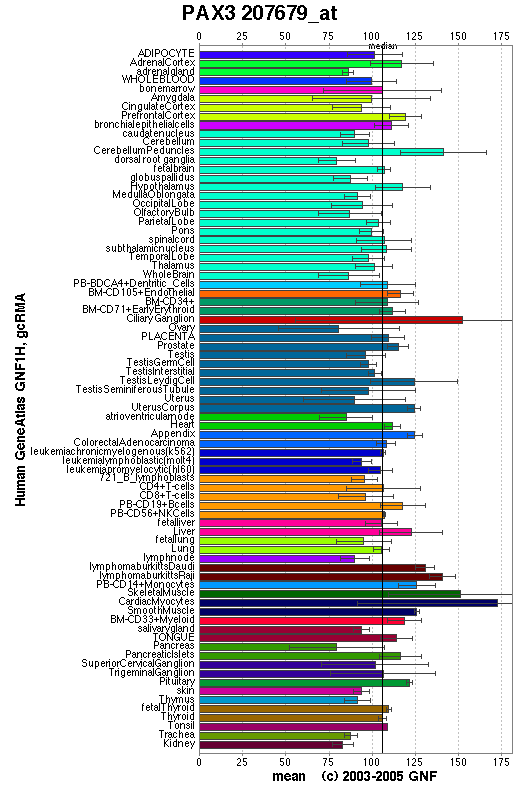
PAX3b is highly expressed in most tissues but PAX3a only in cerebellum, esophagus, and skeletal muscle.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
View more information about PAX3 in Entrez Gene, UCSC Gene Sorter, GeneCards.
PROTEIN
Sequence
a) paired box gene 3 isoform PAX3 ( NP_852122): 479 aa, UniProtKB/Swiss-Prot entry P23760.
b) paired box gene 3 isoform PAX3a (NP_000429): 215 aa, UniProtKB/Swiss-Prot entry Q53T90.
c) paired box gene 3 isoform PAX3b ( NP_039230): 206 aa, UniProtKB/Swiss-Prot entry Q6GSJ9.
d) paired box gene 3 isoform PAX3d ( NP_852123): 484 aa, UniProtKB/Swiss-Prot entry Q494Z3.
e) paired box gene 3 isoform PAX3e ( NP_852124): 505 aa, UniProtKB/Swiss-Prot entry P23760.
f) paired box gene 3 isoform PAX3g ( NP_852126): 403 aa, UniProtKB/Swiss-Prot entry Q86UQ3.
g) paired box gene 3 isoform PAX3h ( NP_852125): 407 aa, UniProtKB/Swiss-Prot entry Q86UQ2.
Ortholog (PAX3e)
Species
Chimpanzee Dog Mouse Rat Chicken GeneView
PAX3
PAX3
Pax3
Pax3
PAX3
Protein
XP_001165390 (505 aa)
XP_545664 (468 aa)
NP_032807 (479 aa)
XP_343602 (476 aa)
NP_989600 (484 aa)
Identities
505/505 (100%) 450/458 (98%) 467/474 (98%) 461/476 (96%) 468/484 (96%)
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc. View evolutionary tree by TreeView.
Domain (PAX3e)
(1) Domains predicted by SMART:
a) PAX: 34-159
b) low complexity: 164-185
c) HOX: 219-281
(2) Transmembrane domains predicted by SOSUI:
This amino acid sequence is of a SOLUBLE PROTEIN.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a)Protein kinase C phosphorylation site:
Site : 95 to 97 SIR.
Site : 180 to 182 SEK.
Site : 193 to 195 SER.
Site : 268 to 270 SNR.
Site : 358 to 360 STR.
Site : 430 to 432 SQR.
b) Casein kinase II phosphorylation site:
Site : 110 to 113 TTPD.
Site : 205 to 208 SDID.
Site : 209 to 212 SEPD.
Site : 248 to 251 TREE.
Site : 346 to 349 SNPD.
Site : 365 to 368 SYTD.
Site : 496 to 499 SSGE.
Site : 497 to 500 SGEE.
c) Tyrosine kinase phosphorylation site:
Site : 236 to 243 RAFERTHY.
d) N-myristoylation site:
Site : 43 to 48 GVFING.
Site : 66 to 71 GIRPCV.
Site : 99 to 104 GAIGGS.
Site : 394 to 399 GLLTNH.
Site : 418 to 423 GLEPTT.
e) 'Homeobox' domain signature:
Site : 252 to 275 LAQRAKLTEARVQVWFSNRRARWR.
f) Paired domain signature.:
Site : 68 to 84 RPCVISRQLRVSHGCVS.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) Tentative number of TMS(s) for the threshold 0.5: 0
c) KDEL ER retention motif in C-terminus: none
d) ER membrane retention signals: none
e) VAC possible vacuolar targeting motif: none
f) Actinin-type actin-binding motif: type 1: none; type 2: none
g) Prenylation motif: none
h) memYQRL transport motif from cell surface to Golgi: none
i) Tyrosines in the tail: none
j) Dileucine motif in the tail: none
3D Model
(1)
ModBase predicted 3D structure from UCSC Genome Sorter:
P23760 ( isoform PAX3 ):
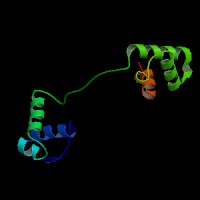 From left to right: Front, Top, and Side views of predicted protein.
From left to right: Front, Top, and Side views of predicted protein.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=55,974.7Da, pI=7.67 (PAX3e).
FUNCTION
Ontology
(1) Biological process: DNA-dependent regulation of transcription, neural crest cell migration, neural tube closure, sensory perception of sound.
(2) Transcription factor activity.
(3) Sequence-specific DNA binding.
(4) Embryogenesis and oncogenesis.
Location
Nucleus.
Interaction
It can bind to DNA as a heterodimer with PAX7. Interacts with DAXX.
Interactions in HPRD.
View interacting proteins in IntAct .
View co-occured partners in literature searched by PPI Finder.
Pathway
PAX3 is involved in RNA polymerase II transcription factor activity. PAX3 synergistically transactivates the promoter of Mitf
with SOX10. Mitf protein in turn regulates the tyrosinase and c-KIT gene.
MUTATION
Allele or SNP
65 mutations deposited in HGMD.
SNPs deposited in dbSNP.
Selected allelic examples described in OMIM.
Distribution
Except the mutations deposited in HGMD, novel mutations are listed here.
Qin, et al (2006) reported a novel mutation in a Chinese WS1 family, c.701T>C (p.L234P).
Kujat, et al (2007) identified a novel splice site mutation within the PAX3 gene in intron 5 in a European WS1 family with spina bifida.
Yang, et al (2007) reported a novel nonsense mutation S209X
in a Chinese WS1 family.
Nakamura, et al (2008) reported a novel mutation in a Japanese WS1 patient, c.801T>A (p.F267I).
Note that the numbering of the mutations in HGMD is based on isoform PAX3 cDNA (NM_181457), view ORF here.
Effect
Most of the mutations in PAX3 are missense/nonsense and small insertion/deletion mutations in WS1 or WS3. To date, 40 missense/nonsense mutations have been reported, 18 mutations reside in the paired domain, and 15 mutations exist in the homeobox domain. In total, 33 out of the 40 reported mutations reside in the DNA binding domains (the paired domain or homeobox domain) of the PAX3 (Nakamura, et al). Chromosomal translocation with PAX3 can cause fusion oncogenic proteins such as PAX3-FKHR, NCOA1-PAX3.
PHENOTYPE
Defects in PAX3 are the cause of Waardenburg syndrome type 1 (WS1) [OMIM:193500]. WS1 is an autosomal dominant disorder characterized by wide bridge of nose owing to lateral displacement of the inner canthus of each eye (dystopia canthorum), pigmentary disturbances such as frontal white blaze of hair, heterochromia of irides, white eyelashes, leukoderma and sensorineural deafness. The syndrome shows variable clinical expression and some affected individuals do not manifest hearing impairment.
Defects in PAX3 are the cause of Waardenburg syndrome type 3 (WS3) [OMIM:148820]; also known as Klein-Waardenburg syndrome or Waardenburg syndrome with upper limb anomalies or white forelock with malformations. WS3 is a very rare autosomal dominant disorder, which shares many of the characteristics of WS1. Patients additionally present with musculoskeletal abnormalities.
Defects in PAX3 are the cause of craniofacial-deafness-hand syndrome (CDHS) [OMIM:122880]. CDHS is thought to be an autosomal dominant disease which comprises absence or hypoplasia of the nasal bones, hypoplastic maxilla, small and short nose with thin nares, limited movement of the wrist, short palpebral fissures, ulnar deviation of the fingers, hypertelorism and profound sensory-neural deafness.
Rhabdomyosarcoma is the most common soft tissue carcinoma in childhood, representing 5-8% of all malignancies in children. Two main histopathologic variants have been described, embryonal (ERMS) and alveolar (ARMS). A chromosomal aberration involving PAX3 is a cause of rhabdomyosarcoma 2 (RMS2) [OMIM:268220]; also known as ARMS. Translocation (2;13)(q35;q14) with FOXO1 produces a fusion protein which is a transcriptional activator.The t(2;13)(q35;q14) and the variant t(1;13)(p36;q14) are seen in a majority of ARMS cases, which are associated with PAX3-FKHR and PAX7-FKHR fusion transcripts, respectively. Translocation t(2;2)(q35;p23) with NCOA1 generates the NCOA1-PAX3 oncogene consisting of the N-terminus part of PAX3 and the C-terminus part of NCOA1. The fusion protein acts as a transcriptional activator (Wachtel, et al). A novel chromosomal t(2;20)(q35;p12) occurring in a case of ERMS (Ho, et al).
REFERENCE
- Ho RH, Johnson J, Dev VG, Whitlock JA. A novel t(2;20)(q35;p12) in embryonal rhabdomyosarcoma. Cancer Genet Cytogenet 2004; 151:73-7. PMID: 15120913
- Kujat A, Veith VP, Faber R, Froster UG. Prenatal diagnosis and genetic counseling in a case of spina bifida in a family with Waardenburg syndrome type I. Fetal Diagn Ther 2007; 22:155-8. PMID: 17139175
- Nakamura M, Ishikawa O, Tokura Y. A novel missense mutation in the PAX3 gene in a case of Waardenburg syndrome type I. J Eur Acad Dermatol Venereol 2008; [Epub ahead of print] [No abstract available]. PMID: 18761541
- Qin W, Shu A, Qian X, Gao J, Xing Q, Zhang J, Zheng Y, Li X, Li S, Feng G, He L.
A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome. Mol Vis 2006; 12:1001-8.
PMID: 16971891
- Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, Buhlmann P, Niggli FK, Schafer BW. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res 2004; 64:5539-45. PMID: 15313887
- Yang SZ, Cao JY, Zhang RN, Liu LX, Liu X, Zhang X, Kang DY, Li M, Han DY, Yuan HJ, Yang WY. Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients. Chin Med J (Engl) 2007; 120:46-9. PMID: 17254487
| Species | Chimpanzee | Dog | Mouse | Rat | Chicken |
| GeneView | PAX3 | PAX3 | Pax3 | Pax3 | PAX3 |
| Protein | XP_001165390 (505 aa) | XP_545664 (468 aa) | NP_032807 (479 aa) | XP_343602 (476 aa) | NP_989600 (484 aa) |
| Identities | 505/505 (100%) | 450/458 (98%) | 467/474 (98%) | 461/476 (96%) | 468/484 (96%) |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc. View evolutionary tree by TreeView.
Domain (PAX3e)
(1) Domains predicted by SMART:
a) PAX: 34-159
b) low complexity: 164-185
c) HOX: 219-281
(2) Transmembrane domains predicted by SOSUI:
This amino acid sequence is of a SOLUBLE PROTEIN.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a)Protein kinase C phosphorylation site:
Site : 95 to 97 SIR.
Site : 180 to 182 SEK.
Site : 193 to 195 SER.
Site : 268 to 270 SNR.
Site : 358 to 360 STR.
Site : 430 to 432 SQR.
b) Casein kinase II phosphorylation site:
Site : 110 to 113 TTPD.
Site : 205 to 208 SDID.
Site : 209 to 212 SEPD.
Site : 248 to 251 TREE.
Site : 346 to 349 SNPD.
Site : 365 to 368 SYTD.
Site : 496 to 499 SSGE.
Site : 497 to 500 SGEE.
c) Tyrosine kinase phosphorylation site:
Site : 236 to 243 RAFERTHY.
d) N-myristoylation site:
Site : 43 to 48 GVFING.
Site : 66 to 71 GIRPCV.
Site : 99 to 104 GAIGGS.
Site : 394 to 399 GLLTNH.
Site : 418 to 423 GLEPTT.
e) 'Homeobox' domain signature:
Site : 252 to 275 LAQRAKLTEARVQVWFSNRRARWR.
f) Paired domain signature.:
Site : 68 to 84 RPCVISRQLRVSHGCVS.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) Tentative number of TMS(s) for the threshold 0.5: 0
c) KDEL ER retention motif in C-terminus: none
d) ER membrane retention signals: none
e) VAC possible vacuolar targeting motif: none
f) Actinin-type actin-binding motif: type 1: none; type 2: none
g) Prenylation motif: none
h) memYQRL transport motif from cell surface to Golgi: none
i) Tyrosines in the tail: none
j) Dileucine motif in the tail: none
3D Model
(1)
ModBase predicted 3D structure from UCSC Genome Sorter:
P23760 ( isoform PAX3 ):
From left to right: Front, Top, and Side views of predicted protein.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=55,974.7Da, pI=7.67 (PAX3e).
(1) Domains predicted by SMART:
a) PAX: 34-159
b) low complexity: 164-185
c) HOX: 219-281
(2) Transmembrane domains predicted by SOSUI:
This amino acid sequence is of a SOLUBLE PROTEIN.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a)Protein kinase C phosphorylation site:
Site : 95 to 97 SIR.
Site : 180 to 182 SEK.
Site : 193 to 195 SER.
Site : 268 to 270 SNR.
Site : 358 to 360 STR.
Site : 430 to 432 SQR.
b) Casein kinase II phosphorylation site:
Site : 110 to 113 TTPD.
Site : 205 to 208 SDID.
Site : 209 to 212 SEPD.
Site : 248 to 251 TREE.
Site : 346 to 349 SNPD.
Site : 365 to 368 SYTD.
Site : 496 to 499 SSGE.
Site : 497 to 500 SGEE.
c) Tyrosine kinase phosphorylation site:
Site : 236 to 243 RAFERTHY.
d) N-myristoylation site:
Site : 43 to 48 GVFING.
Site : 66 to 71 GIRPCV.
Site : 99 to 104 GAIGGS.
Site : 394 to 399 GLLTNH.
Site : 418 to 423 GLEPTT.
e) 'Homeobox' domain signature:
Site : 252 to 275 LAQRAKLTEARVQVWFSNRRARWR.
f) Paired domain signature.:
Site : 68 to 84 RPCVISRQLRVSHGCVS.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) Tentative number of TMS(s) for the threshold 0.5: 0
c) KDEL ER retention motif in C-terminus: none
d) ER membrane retention signals: none
e) VAC possible vacuolar targeting motif: none
f) Actinin-type actin-binding motif: type 1: none; type 2: none
g) Prenylation motif: none
h) memYQRL transport motif from cell surface to Golgi: none
i) Tyrosines in the tail: none
j) Dileucine motif in the tail: none
3D Model
(1) ModBase predicted 3D structure from UCSC Genome Sorter:
P23760 ( isoform PAX3 ):
From left to right: Front, Top, and Side views of predicted protein.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=55,974.7Da, pI=7.67 (PAX3e).
FUNCTION
Ontology
(1) Biological process: DNA-dependent regulation of transcription, neural crest cell migration, neural tube closure, sensory perception of sound.
(2) Transcription factor activity.
(3) Sequence-specific DNA binding.
(4) Embryogenesis and oncogenesis.
Location
Nucleus.
Interaction
It can bind to DNA as a heterodimer with PAX7. Interacts with DAXX.
Interactions in HPRD. View interacting proteins in IntAct .
View co-occured partners in literature searched by PPI Finder.
Pathway
PAX3 is involved in RNA polymerase II transcription factor activity. PAX3 synergistically transactivates the promoter of Mitf with SOX10. Mitf protein in turn regulates the tyrosinase and c-KIT gene.
MUTATION
Allele or SNP
65 mutations deposited in HGMD.
SNPs deposited in dbSNP.
Selected allelic examples described in OMIM.
Distribution
Except the mutations deposited in HGMD, novel mutations are listed here.
Qin, et al (2006) reported a novel mutation in a Chinese WS1 family, c.701T>C (p.L234P).
Kujat, et al (2007) identified a novel splice site mutation within the PAX3 gene in intron 5 in a European WS1 family with spina bifida.
Yang, et al (2007) reported a novel nonsense mutation S209X in a Chinese WS1 family.
Nakamura, et al (2008) reported a novel mutation in a Japanese WS1 patient, c.801T>A (p.F267I).
Note that the numbering of the mutations in HGMD is based on isoform PAX3 cDNA (NM_181457), view ORF here.
Effect
Most of the mutations in PAX3 are missense/nonsense and small insertion/deletion mutations in WS1 or WS3. To date, 40 missense/nonsense mutations have been reported, 18 mutations reside in the paired domain, and 15 mutations exist in the homeobox domain. In total, 33 out of the 40 reported mutations reside in the DNA binding domains (the paired domain or homeobox domain) of the PAX3 (Nakamura, et al). Chromosomal translocation with PAX3 can cause fusion oncogenic proteins such as PAX3-FKHR, NCOA1-PAX3.
PHENOTYPE
Defects in PAX3 are the cause of Waardenburg syndrome type 1 (WS1) [OMIM:193500]. WS1 is an autosomal dominant disorder characterized by wide bridge of nose owing to lateral displacement of the inner canthus of each eye (dystopia canthorum), pigmentary disturbances such as frontal white blaze of hair, heterochromia of irides, white eyelashes, leukoderma and sensorineural deafness. The syndrome shows variable clinical expression and some affected individuals do not manifest hearing impairment.
Defects in PAX3 are the cause of Waardenburg syndrome type 3 (WS3) [OMIM:148820]; also known as Klein-Waardenburg syndrome or Waardenburg syndrome with upper limb anomalies or white forelock with malformations. WS3 is a very rare autosomal dominant disorder, which shares many of the characteristics of WS1. Patients additionally present with musculoskeletal abnormalities.
Defects in PAX3 are the cause of craniofacial-deafness-hand syndrome (CDHS) [OMIM:122880]. CDHS is thought to be an autosomal dominant disease which comprises absence or hypoplasia of the nasal bones, hypoplastic maxilla, small and short nose with thin nares, limited movement of the wrist, short palpebral fissures, ulnar deviation of the fingers, hypertelorism and profound sensory-neural deafness.
Rhabdomyosarcoma is the most common soft tissue carcinoma in childhood, representing 5-8% of all malignancies in children. Two main histopathologic variants have been described, embryonal (ERMS) and alveolar (ARMS). A chromosomal aberration involving PAX3 is a cause of rhabdomyosarcoma 2 (RMS2) [OMIM:268220]; also known as ARMS. Translocation (2;13)(q35;q14) with FOXO1 produces a fusion protein which is a transcriptional activator.The t(2;13)(q35;q14) and the variant t(1;13)(p36;q14) are seen in a majority of ARMS cases, which are associated with PAX3-FKHR and PAX7-FKHR fusion transcripts, respectively. Translocation t(2;2)(q35;p23) with NCOA1 generates the NCOA1-PAX3 oncogene consisting of the N-terminus part of PAX3 and the C-terminus part of NCOA1. The fusion protein acts as a transcriptional activator (Wachtel, et al). A novel chromosomal t(2;20)(q35;p12) occurring in a case of ERMS (Ho, et al).
REFERENCE
- Ho RH, Johnson J, Dev VG, Whitlock JA. A novel t(2;20)(q35;p12) in embryonal rhabdomyosarcoma. Cancer Genet Cytogenet 2004; 151:73-7. PMID: 15120913
- Kujat A, Veith VP, Faber R, Froster UG. Prenatal diagnosis and genetic counseling in a case of spina bifida in a family with Waardenburg syndrome type I. Fetal Diagn Ther 2007; 22:155-8. PMID: 17139175
- Nakamura M, Ishikawa O, Tokura Y. A novel missense mutation in the PAX3 gene in a case of Waardenburg syndrome type I. J Eur Acad Dermatol Venereol 2008; [Epub ahead of print] [No abstract available]. PMID: 18761541
- Qin W, Shu A, Qian X, Gao J, Xing Q, Zhang J, Zheng Y, Li X, Li S, Feng G, He L. A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome. Mol Vis 2006; 12:1001-8. PMID: 16971891
- Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, Buhlmann P, Niggli FK, Schafer BW. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res 2004; 64:5539-45. PMID: 15313887
- Yang SZ, Cao JY, Zhang RN, Liu LX, Liu X, Zhang X, Kang DY, Li M, Han DY, Yuan HJ, Yang WY. Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients. Chin Med J (Engl) 2007; 120:46-9. PMID: 17254487